How FDA is already requiring companies to study the safety and effectiveness of their approved drugs in diverse populations
In recent months, the FDA has released new policies indicating that it will soon require companies to submit plans detailing how they intend to improve the racial and ethnic diversity of their clinical studies prior to approval. But new research from AgencyIQ shows how the FDA is prepared to impose requirements and commitments on companies when they fail to submit sufficient data. Our research shows the range of these requirements, as well as other data that could be useful to sponsors and policymakers alike.
Context: FDA’s recent policy development to increase diversity in clinical trials
- The FDA has long been interested in the diversity of patients enrolled in clinical studies used to support approval. The agency first issued guidance on demographic subgroup analyses in the 1980s. In the intervening years, FDA continued to update guidance in line with updates from the Office of Management and Budget (OMB) and Congressional directives. [ See AgencyIQ’s analysis for more detailed history of FDA’s policy and guidance on diversity in research studies].
- But in recent years, FDA’s efforts to promote and enforce clinical trial diversity have intensified. Most notably, in April 2022 the FDA issued a draft guidance document on diversity in clinical research programs. At a high level, the guidance laid out a policy whereby sponsors of certain products would voluntarily submit a Race and Ethnicity Diversity Plan as part of their development program that would outline and justify their approach to recruiting and retaining a “representative” research population. [ Read AgencyIQ’s extensive analysis of the draft guidance here.]
- One month before, the FDA also put industry on notice when it issued a Complete Response Letter (CRL) to Eli Lilly’s sintilimab after the regulator determined that the sponsor’s data – collected from trial sites in China – was not reflective of the U.S. population the drug was intended to treat. [ Read AgencyIQ’s analysis of the CRL here.]
- In late 2022, reforms within the 2023 Consolidated Appropriations Act gave FDA new statutory authority to require what the law now referred to as Diversity Action Plans (DAPs) for certain premarket clinical studies. Section 3601 of the law amended the Federal Food, Drug and Cosmetic (FD&C) Act to expressly allow FDA to require submission of a DAP for certain Phase 3 and pivotal studies of medical products, including drugs. According to the law, the DAP is a plan, developed by the product sponsor, that would need to include specific enrollment goals, the rationale by which these goals were developed, and “an explanation of how the sponsor intends to meet such goals.”
- The law also directed the FDA to “update or issue guidance” on DAPs by the end of December 2023 – a deadline that the FDA missed. However, the agency issued two new draft guidance documents related to diversity in the meantime in the interim period.
- First, an August 2023 new draft guidance on post-market approaches for obtaining data in underrepresented groups. In other words, if a company did not collect enough data on the use of their products in a specific demographic prior to approval, the FDA might require that it be collected later. The document clarifies that “there are various mechanisms for obtaining postmarketing data on underrepresented populations,” in particular, postmarketing requirements (PMRs), postmarketing commitments (PMCs), or a confirmatory trial for products granted accelerated approval. As AgencyIQ has previously discussed, PMRs are mandatory trials or studies levied by the FDA, while PMCs are post-market studies or trials that are agreed to by the sponsor but are not legally (statutorily or via regulation) required. The guidance explains that in this context, a PMR could be applied if there’s a safety signal, while a PMC could address underrepresentation more broadly. This aligns with the typical use of PMRs and PMCs, in which PMRs focus on known or potential risks, while PMCs can fill in data gaps or bolster safety/efficacy information. [ Read AgencyIQ’s analysis of the 2023 draft guidance here.]
- Second, in January 2024 the FDA updated a 2016 guidance on general collection of race and ethnicity data. The new draft offers recommendations to be used for collecting and reporting this information, which continues to be based on OMB Directive 15. Directive 15 defines the core set of minimum categories of race and ethnicity and offers recommendations on data capture. [ Read AgencyIQ’s full analysis of that draft guidance document here.] A few months later in late March 2024, OMB finalized some long-awaited updates to Directive 15. Following an “ initial set of recommended revisions” from the White House Office of the Chief Statistician in January 2023, Directive 15 has now been formally updated at the federal level. At a high level, this entailed three main changes: (1) race and ethnicity are now combined into one question; (2) Middle Eastern and North African (MENA) has been adopted as a new minimum category; and (3) the Directive now “requires the collection of more detail beyond the minimum required race and ethnicity reporting categories” in most situations. [ Read AgencyIQ’s full analysis of the changes here.]
- The FDA ultimately released its Diversity Action Plan guidance in June 2024. The document offers granular recommendations on how sponsors of both drug and device programs should comply with the new legal requirements, including how to design a DAP and set enrollment goals using data about the incidence and/or prevalence of a disease in the U.S. population. These goals and any adjustments should be explained through a rationale, and a plan must be developed with specific enrollment and retention strategies. Finally, sponsors must articulate a plan to measure progress toward meeting the goals. [Read AgencyIQ’s extensive initial analysis and follow-up here.] The comment period for the draft guidance document is currently open until September 25, 2024. Assuming that the comment period is not extended, and FDA meets its statutory timeline to finalize the guidance within nine months of the closing of the comment period, the guidance could theoretically be put into force by late 2025.
All of this raises an important question: Will FDA leverage its postmarket toolkit for issues related to diversity?
- But the DAP guidance document did not answer to a key question: What happens if targets are not met? As AgencyIQ has previously discussed, the intersection of DAP policy with FDA’s post-market tools is unclear. The 2022 version of the DAP guidance included the following footnote, which was the subject of much discussion in comments on the document: “In the event that recruitment goals are not met despite best efforts, sponsors should discuss with FDA a plan to collect this data in the post-marketing setting” (see footnote 24). However, the new 2024 version of the document does not include any information on the subject, and does not address the question of what happens if studies under DAPs are unable to reach their goals in the pre-market setting.
- However, there is recent precedence for the FDA using PMRs and PMCs to obtain data related to clinical trial diversity, especially for oncology products. For example, several oncology drugs granted accelerated approval in 2023 were required to conduct confirmatory studies and to enroll a study population that sufficiently reflects the racial and ethnic diversity of the U.S. patient population with the disease (See Jaypirca (pirtobrutinib) approval letter and Lunsumio (mosunetuzumab-axgb) approval letter).
- That said, these examples are anecdotal, and AgencyIQ wanted to know the extent to which FDA relied on this sort of approach more systematically. This led AgencyIQ to carry out a comprehensive assessment of the extent to which FDA has asked companies to address diversity-related concerns in a postmarket setting and what FDA wants from those same studies.
Before we talk about the results of our analysis on post-marketing diversity requirements, let’s first discuss our methods.
- The following analysis is based on FDA’s Downloadable Database of Postmarketing Requirements and Commitments, which was last updated July 31, 2024. Altogether, the database comprises over 2,500 unique PMRs and PMCs. According to FDA, the dataset contains all open PMRs and 506B PMCs (those with annual reporting requirements), as well as those have been closed within the last year due to fulfillment or release. This means that the following analysis excludes requirements or commitments closed prior to late July 2023. Of note, AgencyIQ also excluded entries for products with approvals that have been withdrawn.
- To curate this pool to PMRs and PMCs related to diversity, AgencyIQ carried out a two-step filtration process. First, the dataset was limited to entries for which the text of the requirement or commitment contained keywords related to racial and ethnic diversity or representativeness of the population to be studied. Specifically, these entries contained the words diverse, diversity, representative, underrepresented reflective, minority or demographic. In addition, a search was performed for the race and ethnicity categories within OMB Directive 15. The scope of this analysis did not include additional parameters of diversity often recognized by the agency as pertaining to diversity such as sex, age, and socioeconomic status.
- Second, these entries were narrowed down to those with an intentional element related to diversity. In other words, we only wanted to find cases where FDA asked the sponsors to go further than simply collecting demographic information.
- Specifically, PMR and PMC entries were included in the analysis set if the task had one of two of the following characteristics. First, FDA specifically asks the sponsor to ensure a representative population. For example, PMC 4248-1 for AstraZeneca’s Lynparza (Olaparib) asks the sponsor to do the following: “Conduct analyses of clinical trial data to characterize the safety and pharmacokinetics of olaparib across a diverse population. To support a comparative assessment across all U.S. race and ethnic populations, ensure that racial and ethnic minorities are sufficiently represented in the analysis. Include a tabular summary of the available pharmacokinetic data by each racial and ethnic group. Provide the specific racial ethnic group as well as the geographic location of each patient.” Second, FDA asks the sponsor to enrich data for underrepresented groups. For example, PMC 4074-3 for Janssen’s Darzalex Faspro (daratumumab and hyaluronidase-fihj) has the following ask: “Submit a final report containing data from clinical trials, post-marketing reports, compassionate use/expanded access programs, real-world evidence, and other sources to further characterize the safety and efficacy of daratumumab (SC) in combination with pomalidomide and dexamethasone among U.S. racial and ethnic minority patients with multiple myeloma.”
- Using this approach, AgencyIQ identified 55 total relevant PMRs and PMCs. The data signals that these PMRs and PMCs have become more common in recent years. The oldest active requirement was issued in 2018 related to the approval of Otsuka Pharmaceutical’s Jynarque. The newest requirement in this group comes from the March 2024 approval of a supplemental application for a new indication for Beigene’s Brukinska.
- The dataset also had a similar distribution of requirements (24) and commitments (31). In addition, there was no specific application type that was observed to be overwhelmingly more likely to receive this type of PMC or PMR. That said, there were more New Drug Applications (NDAs, 29) than Biologic License Applications (26), and more original applications (35) than supplementary applications (20).
What does FDA ask for in its diversity-focused PMRs and PMCs? There are four distinct types of requirements.
- Through examining the text of each relevant PMR and PMC, AgencyIQ identified four distinct types of asks for sponsors (see the Appendix at the end of this piece for the full text of each requirement). In many cases, specific phrasing and descriptions were retained across different products, indicating that the agency leans on a few “template” PMRs and PMCs when seeking post-market study of diversity. In other words, sponsors can expect to see similar language in these types of requirements and commitments.
- First, FDA will frequently asked sponsors to conduct a new clinical trial. In many of these cases, the agency asks for a phase 3, randomized trial to verify and describe clinical benefit or better understand a safety signal. Most of these entries contain some variation of the sentence, “The trial should include sufficient numbers of racial and ethnic minority patients to better reflect the U.S. patient population and allow for interpretation of the results in these patient populations.” However, a few new clinical trials are asked to enrich certain populations. For example, PMR 4106-11 for Kadmon Pharmaceutical’s Rezurock (belumosudil) instructs the firm to, “Conduct a clinical trial in a sufficient number of Black patients with chronic graft versus host disease to assess the risk of cardiac toxicities and further characterize Grade 3 toxicities including gastrointestinal and vascular disorders associated with the use of belumosudil.”
- Second, FDA might ask sponsors to conduct additional clinical data analysis, which won’t necessarily mandate a new trial. For example, Helsinn Healthcare received an Accelerated Approval PMR for Truseltiq (Infigratinib) that instructed the firm to: “Submit the final progression-free survival (as assessed by blinded independent review) analysis and interim overall survival analysis at the time of final progression-free survival analysis, including datasets from a randomized clinical trial comparing infigratinib to chemotherapy to verify and describe the clinical benefit of infigratinib in patients with advanced or metastatic cholangiocarcinoma harboring an FGFR2 gene fusion or other rearrangement. Ensure that racial and ethnic minority subjects are adequately represented in the trial population, at a minimum, proportional to the prevalence of FGFR2 alterations in these subgroups in the US population.”
- Third, FDA often asked sponsors to conduct what it referred to as an “integrated analysis.” In general, these are PMCs that task the sponsor with using “data from clinical trials and other data sources such as post-marketing reports, real-world evidence and other sources” to answer a question in an underrepresented population with the disease or condition. These commitments also typically specify that the integrated analysis should “support comparative efficacy and safety analyses” between the underrepresented population and majority group.
- Finally, the agency asked for a prospective observational study in rare cases. For example, AbbVie’s Oriahnn (elagolix, estradiol and norethindrone acetate) received PMR 3837-3, which sought “a prospective observational study in premenopausal women receiving treatment with Oriahnn to assess the incidence rate, time to onset, pattern, extent, and reversibility of alopecia, as well as any racial/ethnic differences in developing alopecia.”

- According to AgencyIQ’s analysis, FDA allotted different timelines for sponsors to complete various types of PMRs and PMCs. When comparing the timeframes between assignment of a given requirement or commitment (not the original application approval) and the agency’s original final report due date, it becomes apparent that clinical data analysis tasks generally have a shorter time frame than the more intensive integrated analyses or new clinical trials. While only two such entries were in the analysis set, prospective observational trials were given additional time to complete.
| Category | Average timeframe (years) | Median timeframe (years) | Minimum timeframe (years) | Maximum timeframe (years) |
| New clinical trial | 4.55 | 4.69 | 1.46 | 7.13 |
| Clinical data analysis | 4.05 | 4.57 | 1.94 | 6.09 |
| Integrated analysis | 5.39 | 5.32 | 3.89 | 6.84 |
| Prospective observational study | 7.31 | – | 7.02 | 7.59 |
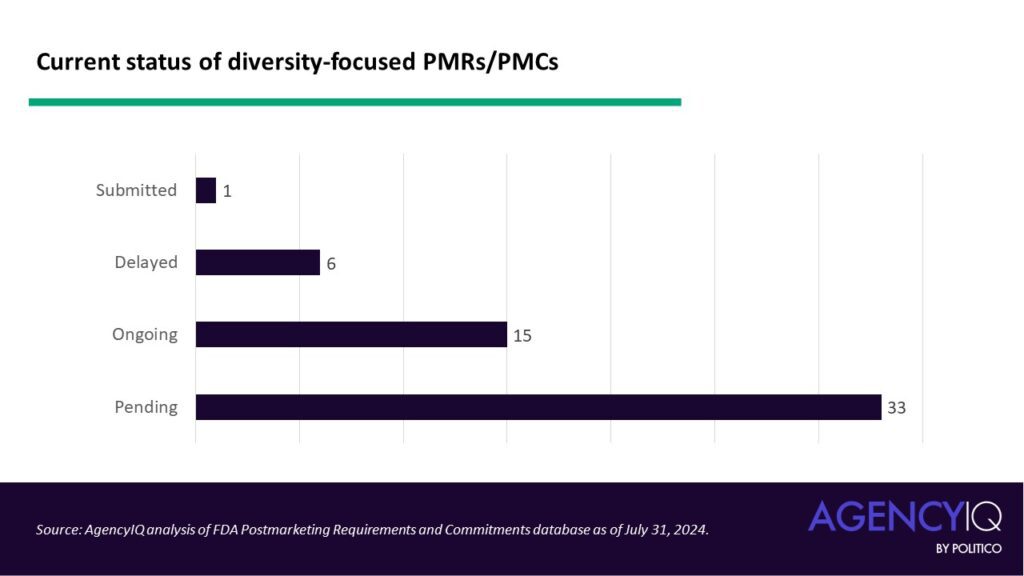
The status: most diversity-focused PMRs and PMCs are currently pending
- FDA’s database contains information pertaining to the status of the open PMRs and PMCs at the time of the update (for our analysis, late July 2024). The status is reported in the following categories, as defined on FDA’s webpage.
|
Status |
Definition |
|
Pending |
The study has not been initiated (i.e., no subjects have been enrolled or animals dosed) but does not meet the criterion for delayed. |
|
Ongoing |
The study is proceeding according to, or is ahead of, the original schedule. |
|
Delayed |
The progression of the study is behind the original study schedule. |
|
Submitted |
The applicant has concluded or terminated the study and has submitted a final study report to the FDA. |
|
Fulfilled |
Upon review of the final study report, FDA is satisfied that the applicant has met the terms of the commitment. |
|
Terminated |
The applicant ended the study before completion and has not yet submitted a final study report to the FDA. |
|
Released |
FDA has informed the applicant that it has been released from its obligation to conduct the postmarketing study because the study is either no longer feasible or would no longer provide useful information. |
- Overall, 60% of the diversity-focused PMRs and PMCs are currently pending, meaning they have yet to be initiated. Just one entry in the dataset has been submitted to the agency, with the rest proceeding either on or off the original study schedule.
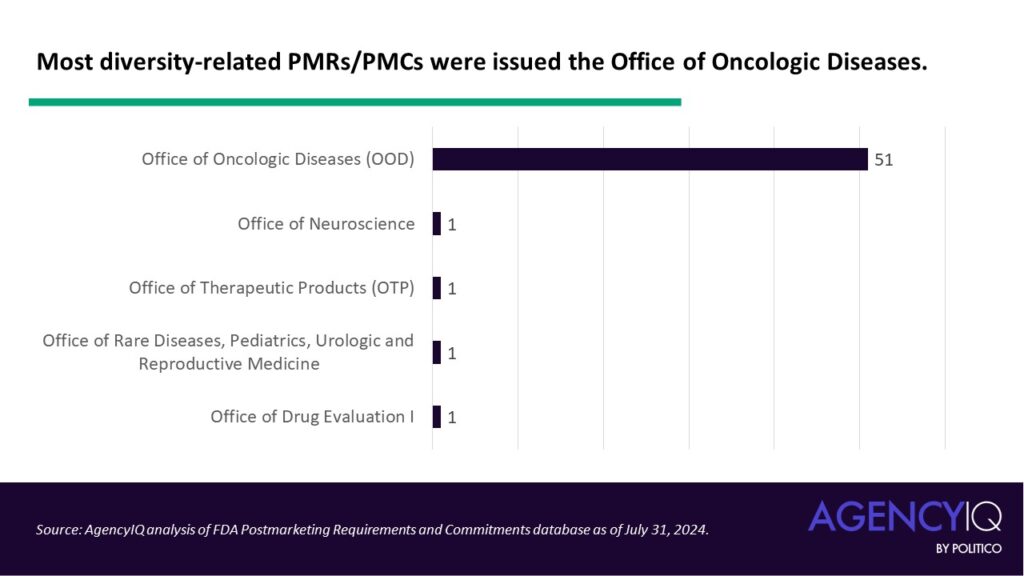
The assigners: Just one FDA review office has issued the overwhelming majority of diversity-focused PMRs and PMCs
- The majority of diversity-related PMRs/PMCs were issued by FDA’s Office of Oncologic Diseases (OOD). Other offices that issued these types of PMCs or PMRs were the Office of Neuroscience, Office of Therapeutic Products, Office of Office of Rare Diseases, Pediatrics, Urologic and Reproductive Medicine, and the now-reorganized Office of Drug Evaluation.
- OOD works in partnership with the agency’s Oncology Center of Excellence (OCE). This is important because OCE has functioned as a catalyst for many of FDA’s diversity-oriented policy activities. The Center wrote FDA’s original 2022 draft guidance on voluntary diversity plans. As AgencyIQ has previously discussed, it appears OCE also took the lead with drafting the new DAP guidance document; LOLA FASHOYIN-AJE (previously with OCE, and now at CBER’s Office of Therapeutic Products) is listed as the top point of contact on the guidance, along with TAMY KIM, who is OCE’s director for regulatory affairs and policy.
- Within OOD, most assignments came from the Division of Hematologic Malignancies 2 (DHM2). According to our analysis, requirements and commitments issued by DHM2 make up nearly half (47.37%) of the entire dataset. FDA’s webpage states that DHM2 is charged with reviewing regulatory submissions for products proposed to treat for lymphoma, chronic lymphocytic leukemia, multiple myeloma, and other plasma cell malignancies. The division is led by Director NICOLE GORMLEY. Gormley has had a leadership role in the agency’s diversity initiatives. For example, in May 2024, OCE announced that Gormley would be a co-director of Project Equity’s expansion from “project” to “program” [ Read AgencyIQ’s full analysis here.].

Within OOD, most assignments came from the Division of Hematologic Malignancies 2
Analysis
- These data indicate how the FDA is already making use of postmarketing commitments, and what drug companies and public stakeholders can expect. This data represents a starting point; as FDA’s new policies for clinical study diversity are implemented in coming years, the volume and makeup of these assignments is likely to change, especially in the wake of the agency’s 2023 draft guidance on obtaining data on underrepresented groups in postmarket settings. They are also likely to change as they are used more outside of the field of oncology.
- It’s worth noting that for “integrated analysis” types of PMCs, the FDA has said that real-world evidence (RWE) is an acceptable source of supportive data. This is notable it may make it easier for some companies to collect additional data without necessarily running clinical studies.
- While FDA’s authority to assign postmarketing studies is solid, industry’s performance with fulfilling these assignments has been historically lackluster. As required under the 1997 Food and Drug Administration Modernization Act (FDAMA), the agency creates a public report that provides a snapshot of sponsor performance on PMRs and PMCs each federal fiscal year (FY). The most recent report covered FY 2022 and was released in late April 2024. In that year, 30% of all required postmarketing studies had not yet been completed and were off schedule for future completion. In addition, just half of all PMRs established in FY 2016 had been fulfilled. Recent legislative reforms to FDA oversight of PMR completion status for accelerated approvals may make an impact on future reports, though it will likely take some time to see an effect reflected in the data.
Featuring previous research by Laura DiAngelo.
To contact the author of this item, please email Amanda Conti ( aconti@agencyiq.com).
To contact the editor of this item, please email Alexander Gaffney ( agaffney@agencyiq.com)
Key Documents and Dates
Appendix: Postmarketing requirements and commitments referencing trial diversity factors
| Applicant Name | Product Name | PMR/PMC Description |
| ABBVIE INC | Oriahnn (elagolix, estradiol and norethindrone acetate) | PMR 3837-3: A prospective observational study in premenopausal women receiving treatment with Oriahnn to assess the incidence rate, time to onset, pattern, extent, and reversibility of alopecia, as well as any racial/ethnic differences in developing alopecia. Physician/observer-reported outcome and/or patient survey should be developed and included in the PMR study to capture timing, pattern, extent, and reversibility of alopecia cases. The study shall evaluate 50 cases of alopecia. |
| ADC Therapeutics SA | Loncastuximab Tesirine | PMR 4029-4: Submit an integrated final report containing data from clinical trials to further characterize the exposure of loncastuximab tesirine-lpyl monotherapy and in combination with immunochemotherapy, the increased risk of severe and serious adverse events, including severe neutropenia, and efficacy among U.S. racial and ethnic minority patients with large B-cell lymphoma. Provide the population pharmacokinetic and exposure-response analyses for both efficacy and safety in the interim report. |
| ADC Therapeutics SA | Loncastuximab Tesirine | PMR 4029-1: Conduct a randomized, phase 3 clinical trial to verify and describe the clinical benefit of loncastuximab tesirine-lpyl in patients with relapsed or refractory large B-cell lymphoma. The trial should include sufficient numbers of racial and ethnic minority patients to better reflect the U.S. patient population and allow for interpretation of the results in these patient populations. Patients should be randomized to receive loncastuximab tesirine-lpyl plus immunotherapy or immunochemotherapy. The primary endpoint should be progression-free survival, with secondary endpoints that include overall survival and objective response rate. |
| ASTRAZENECA PHARMACEUTICALS LP | Lynparza (Olaparib) | PMC 4248-1: Conduct analyses of clinical trial data to characterize the safety and pharmacokinetics of olaparib across a diverse population. To support a comparative assessment across all U.S. race and ethnic populations, ensure that racial and ethnic minorities are sufficiently represented in the analysis. Include a tabular summary of the available pharmacokinetic data by each racial and ethnic group. Provide the specific racial ethnic group as well as the geographic location of each patient. |
| ASTRAZENECA PHARMACEUTICALS LP | Truqap (capivasertib) | PMC 4548-5: Conduct an integrated analysis of data from clinical trials and other data sources such as post-marketing reports, real-world evidence and other sources to further characterize the safety and efficacy of capivasertib in combination with fulvestrant in patients of underrepresented racial and ethnic minority groups with hormone receptor positive, HER2 negative metastatic breast cancer with one or more PIK3CA/AKT1/PTENalterations. The analyses should include safety and efficacy outcome analyses by race and ethnicity. |
| BEIGENE USA INC | BRUKINSA (zanubrutinib) | PMC 4130-2: Conduct a study to further characterize the clinical benefit and safety of zanubrutinib for the treatment of patients with newly diagnosed Waldenström’s Macroglobulinemia with MYD88 mutation. This should include an assessment of the CXCR4 mutation status. In addition, the study should include a sufficient number of patients enrolled in the United States and sufficient numbers of racial and ethnic minority patients to allow for the interpretation of the results in these patient populations. |
| BEIGENE USA INC | BRUKINSA (zanubrutinib) | PMC 4130-3: Conduct a study to further characterize the clinical benefit and safety of zanubrutinib in patients with newly diagnosed and relapsed/refractory Waldenström’s Macroglobulinemia with MYD88wt. This study should include a sufficient number of patients enrolled in the United States and sufficient numbers of racial and ethnic minority patients to allow for the interpretation of the results in these patient populations. |
| BEIGENE USA INC | BRUKINSA (zanubrutinib) | PMC 4130-4: Conduct an integrated analysis containing data from clinical trials and other data sources such as post-marketing reports, real-world evidence and other sources to further characterize the safety and efficacy of zanubrutinib in racial and ethnic minorities with Waldenström’s Macroglobulinemia. |
| BEIGENE USA INC | BRUKINSA (zanubrutinib) | PMR 4128-1: Conduct a randomized clinical trial that verifies and describes the clinical benefit of zanubrutinib in patients with relapsed or refractory marginal zone lymphoma. The trial should include sufficient representation of racial and ethnic minorities to better reflect the U.S. patient population and allow for interpretation of the results in these patient populations. The primary endpoint should be progression-free survival, with secondary endpoints that include objective response rate and overall survival. |
| BEIGENE USA INC | BRUKINSA (zanubrutinib) | PMR 4583-1: Complete a randomized clinical trial that evaluates the clinical benefit of zanubrutinib plus obinutuzumab versus lenalidomide plus rituximab in patients with relapsed or refractory follicular lymphoma. The primary endpoint should be progression-free survival, with secondary endpoints that include objective response rate and overall survival. The trial should enroll a sufficiently representative study population that reflects the racial and ethnic diversity of the U.S. population of patients with follicular lymphoma and that allows for interpretation of the results in these populations. |
| Bristol-Myers Squibb Company | Opdivo (Nivolumab) | PMC 4245-2: Conduct an integrated analysis from postmarketing data sources or ongoing/planned clinical trials enrolling a sufficient representation of adults ages 75 years and older, and United States (U.S.) racial and ethnic minority patients that is reflective of the U.S. population of patients with NSCLC to further characterize the safety and efficacy of nivolumab in combination with platinum-doublet chemotherapy in these patients. In the analysis, include a sufficient number of patients enrolled in the U.S. ages 75 years and older, and a sufficient number of racial and ethnic minorities reflecting of the incidence of NSCLC in each subpopulation to allow for interpretation of the results. |
| Celgene | ABECMA, idecabtagene vicleucel | Celgene commits to submit an integrated final report containing data from clinical trials MM-002 and MM-003 to further characterize the safety and efficacy of idecabtagene vicleucel among African-Americans/ Blacks with multiple myeloma. The primary objective of this analysis is to evaluate the efficacy of idecabtagene vicleucel in the subpopulation of African-Americans/Blacks with multiple myeloma compared to the subpopulation of Whites, and the secondary objective is safety. Ensure that the representation of the African American subpopulation in the studies is reflective of the Black population in the geographical location/country. Therefore, approximately 15% of the population that is enrolled from the US should comprise of African Americans. Prespecify an analysis plan for safety and efficacy with a justification/rationale of prespecified assumptions for efficacy outcomes. |
| Coherus BioSciences, Inc. | Loqtorzi (toripalimab-tpzi) | PMC 4471-1: Conduct a clinical trial enrolling a total sample size of 100 patients in the United States (U.S.) and Canada, that includes a sufficient representation of patients in racial and ethnic minority subgroups and is reflective of the U.S. population of patients with nasopharyngeal carcinoma (NPC), to further characterize the safety and efficacy of toripalimab in combination with cisplatin and gemcitabine in these patients. Include a sufficient number of patients with the keratinizing subtype reflecting the incidence of keratinizing NPC in the U.S. population. Conduct sparse sampling for supportive population pharmacokinetic and Exposure-Response (E-R) analyses. The E-R analyses may be used as supportive evidence for the efficacy and safety in the intended patient population. In the E-R analyses report, include an analysis of the presence and clinical impact of the neutralizing anti-drug antibodies on pharmacokinetics, efficacy and safety of toripalimab. |
| Daiichi Sankyo, Inc. | ENHERTU (fam-trastuzumab deruxtecan-nxki) | PMC 4318-1: Conduct an integrated analysis containing data from clinical trials and other data sources such as post-marketing reports, real-world evidence and other sources to further characterize the safety and efficacy of T-DXd in racial and ethnic minority patients and older patients age >65 years with HER2-low metastatic breast cancer. The analyses should support comparative safety and efficacy outcome analyses between the aforementioned populations and White and younger patients. |
| EPIZYME INC | Tazverik (tazemetostat) | PMC 3872-6: Submit a final report containing data from clinical trials, post-marketing reports, compassionate use/expanded access program, real-world evidence, and other sources to further characterize the safety and efficacy of tazemetostat monotherapy and tazemetostat in combination with other immunotherapy among U.S. racial and ethnic minority patients with follicular lymphoma. |
| EPIZYME INC | Tazverik (tazemetostat) | PMC 3872-6: Submit a final report containing data from clinical trials, post-marketing reports, compassionate use/expanded access program, real-world evidence, and other sources to further characterize the safety and efficacy of tazemetostat monotherapy and tazemetostat in combination with other immunotherapy among U.S. racial and ethnic minority patients with follicular lymphoma. |
| EPIZYME INC | Tazverik (tazemetostat) | PMR 3872-1: Submit the final report and datasets from a randomized, phase 3 clinical trial that verifies and describes the clinical benefit of tazemetostat in patients with relapsed or refractory follicular lymphoma whose tumors are positive for an EZH2 mutation as detected by an FDA-approved test. The trial should include sufficient numbers of racial and ethnic minority patients to better reflect the U.S. patient population and allow for the interpretation of the results in these patient populations. Patients should be randomized to receive immunotherapy with or without tazemetostat. The primary endpoint should be progression-free survival, with secondary endpoints that include overall survival and objective response rate. |
| EPIZYME INC | Tazverik (tazemetostat) | PMR 3872-2: Submit the final report and datasets from a randomized, phase 3 clinical trial that verifies and describes the clinical benefit of tazemetostat in patients with relapsed or refractory follicular lymphoma whose tumors do not have an EZH2 mutation as detected by an FDA-approved test. The trial should include sufficient numbers of racial and ethnic minority patients to better reflect the U.S. patient population and allow for the interpretation of the results in these patient populations. Patients should be randomized to receive immunotherapy with or without tazemetostat. The primary endpoint should be progression-free survival, with secondary endpoints that include overall survival and objective response rate. |
| EPIZYME INC | Tazverik (tazemetostat) | PMR 3872-1: Submit the final report and datasets from a randomized, phase 3 clinical trial that verifies and describes the clinical benefit of tazemetostat in patients with relapsed or refractory follicular lymphoma whose tumors are positive for an EZH2 mutation as detected by an FDA-approved test. The trial should include sufficient numbers of racial and ethnic minority patients to better reflect the U.S. patient population and allow for the interpretation of the results in these patient populations. Patients should be randomized to receive immunotherapy with or without tazemetostat. The primary endpoint should be progression-free survival, with secondary endpoints that include overall survival and objective response rate. |
| EPIZYME INC | Tazverik (tazemetostat) | PMR 3872-2: Submit the final report and datasets from a randomized, phase 3 clinical trial that verifies and describes the clinical benefit of tazemetostat in patients with relapsed or refractory follicular lymphoma whose tumors do not have an EZH2 mutation as detected by an FDA-approved test. The trial should include sufficient numbers of racial and ethnic minority patients to better reflect the U.S. patient population and allow for the interpretation of the results in these patient populations. Patients should be randomized to receive immunotherapy with or without tazemetostat. The primary endpoint should be progression-free survival, with secondary endpoints that include overall survival and objective response rate. |
| FABRE KRAMER PHARMACEUTICALS INC | Exxua (gepirone) | PMC 4485-5: Conduct a controlled trial to evaluate the longer-term (i.e., maintenance) efficacy of gepirone ER in the treatment of adults with major depressive disorder (MDD). The population should include significant U.S. representation and include underrepresented racial and ethnic minorities. This trial must include a placebo group and must utilize a double-blind, randomized-withdrawal design following an adequate period of stabilization with open-label treatment of gepirone ER. This trial design must incorporate long-term safety assessments (including pre-, post-, and on-treatment electrocardiograms). Because the short-term trials used to support efficacy were not fixed-dose, it is important to establish the dose-response for maintenance. Therefore, following open-label stabilization, this trial should randomize subjects to fixed doses of the to-be-marketed doses of gepirone ER (and placebo) during the maintenance phase. |
| Genentech, Inc. | Columvi (glofitamab-gxbm) | PMC 4464-3: Conduct an integrated analysis of data from clinical trials to further characterize the safety, efficacy, pharmacokinetics, and pharmacodynamics of glofitamab among U.S. racial and ethnic minority patients with large B-cell lymphoma. The population should be representative of the U.S. population of patients with large B-cell lymphoma, including racial and ethnic diversity, and allow for interpretation of the results in these populations. |
| Genentech, Inc. | Lunsumio (mosunetuzumab-axgb) | PMR 4375-1: Conduct a randomized clinical trial in patients with relapsed or refractory follicular lymphoma, with patients randomized to receive mosunetuzumab in combination with lenalidomide or rituximab in combination with lenalidomide. The primary endpoint should be progression-free survival, with secondary endpoints that include response rate and overall survival. The trial should enroll a sufficiently representative study population to reflect the racial and ethnic diversity of the U.S. patient population with follicular lymphoma and allow for interpretation of the results in these patient populations. |
| Genmab US, Inc. | Epkinly (epcoritamab-bysp) | PMC 4435-4: Conduct an integrated analysis of data from clinical trials to further characterize the safety, efficacy, pharmacokinetics, and pharmacodynamics of epcoritamab monotherapy among U.S. racial and ethnic minority patients with large B-cell lymphoma. The population should be representative of the U.S. population, including racial and ethnic diversity, of patients with large B-cell lymphoma and allow for interpretation of the results in these populations. |
| GlaxoSmithKline LLC | Jemperli (dostarlimab-gxly) | PMC 4404-1: Conduct an integrated analysis containing data from clinical trials and other data sources such as post-marketing reports, real-world evidence and other sources to further characterize the efficacy and safety of dostarlimab in patients with dMMR endometrial cancer from racial and ethnic minority groups. The analyses should support comparative efficacy and safety analyses between the aforementioned populations and White patients. |
| HELSINN HEALTHCARE SA | Truseltiq (Infigratinib) | PMR 4067-1: Submit the final progression-free survival (as assessed by blinded independent review) analysis and interim overall survival analysis at the time of final progression-free survival analysis, including datasets from a randomized clinical trial comparing infigratinib to chemotherapy to verify and describe the clinical benefit of infigratinib in patients with advanced or metastatic cholangiocarcinoma harboring an FGFR2 gene fusion or other rearrangement. Ensure that racial and ethnic minority subjects are adequately represented in the trial population, at a minimum, proportiona to the prevalence of FGFR2 alterations in these subgroups in the US population. |
| HELSINN HEALTHCARE SA | Truseltiq (Infigratinib) | PMR 4067-4: Conduct a clinical trial to further characterize the serious adverse reactions of hyperphosphatemia and eye disorders in patients with firstline or refractory cholangiocarcinoma harboring an FGFR2 fusion or other rearrangement receiving alternate dosage(s) regimens of infigratinib. Characterize all serious adverse events including hyperphosphatemia and eye disorders, dose reductions, interruptions, and discontinuations due to serious adverse events. Compare clinical efficacy and safety descriptively across concurrently-enrolled, parallel cohorts evaluating the approved infigratinib dosage and an alternate dosage regimen. Include sparse PK samples for exposure-response analyses for efficacy and safety and conduct exploratory PK/PD analysis using serum phosphate levels. Ensure that racial and ethnic minority subjects are adequately represented in the trial population, at a minimum, proportional to the prevalence of FGFR2 alterations in these subgroups in the US population. |
| ImmunoGen, Inc. | Elahere (mirvetuximab soravtansine-gynx) | PMC 4347-5: Conduct an integrated analysis containing data from clinical trials and other data sources such as post-marketing reports, real-world evidence and other sources to further characterize the safety and efficacy of mirvetuximab soravtansine in patients from racial and ethnic minority groups. The analyses should support comparative safety and efficacy outcome analyses between the aforementioned populations and White patients. |
| Janssen Biotech, Inc. | RYBREVANT (amivantamab-vmjw) | PMC 4070-3: Submit a final report containing data from clinical trials enrolling a sufficient representation of Black or African American patients that is reflective of the U.S. population of patients with EGFR exon 20 insertion mutated NSCLC to further characterize the safety and efficacy of amivantamab-vmjw in Black or African American patients with EGFR exon 20 insertion mutated NSCLC. |
| Janssen Biotech, Inc. | Darzalex Faspro (daratumumab and hyaluronidase-fihj) | PMR 4074-1: Conduct a clinical study to further characterize the exposure of daratumumab (D) subcutaneous (SC), the increased risk of severe and serious adverse events, including severe neutropenia, and efficacy among U.S. racial and ethnic minority patients with relapsed or refractory multiple myeloma. Include an assessment of the PK, PD, safety, and efficacy of daratumumab SC in combination with other agents including pomalidomide and dexamethasone (Pd) in U.S. racial and ethnic minority patients including Black and Asian patients with relapsed or refractory multiple myeloma in the final study report. The population pharmacokinetic and exposure-response analyses for both efficacy and safety should be updated. |
| Janssen Biotech, Inc. | Darzalex Faspro (daratumumab and hyaluronidase-fihj) | PMC 4074-3: Submit a final report containing data from clinical trials, post-marketing reports, compassionate use/expanded access programs, real-world evidence, and other sources to further characterize the safety and efficacy of daratumumab (SC) in combination with pomalidomide and dexamethasone among U.S. racial and ethnic minority patients with multiple myeloma. |
| Janssen Biotech, Inc. | Darzalex Faspro (daratumumab and hyaluronidase-fihj) | PMC 4183-1: Conduct an integrated study analysis containing data from clinical trials, post-marketing reports, compassionate use/expanded access programs, real-world evidence, and other sources to further characterize the safety and efficacy of daratumumab (SC) in combination with carfilzomib and dexamethasone among U.S. racial and ethnic minority patients with multiple myeloma. |
| Janssen Biotech, Inc. | RYBREVANT (amivantamab-vmjw) | PMC 4597-2: Conduct an integrated analysis containing data from clinical trials and other data sources, such as real-world evidence and post-marketing clinical trial reports, to further characterize the efficacy and safety of amivantamab in combination with carboplatin and pemetrexed in patients ages 75 years and older, Black or African American patients, and patients of Latino ethnicity, diagnosed with metastatic NSCLC harboring EGFR exon 20 insertion mutations. |
| Janssen Biotech, Inc. | Darzalex Faspro (daratumumab and hyaluronidase-fihj) | PMR 3951-3: Conduct a clinical trial to assess the safety of daratumumab subcutaneous (SC) among U.S. racial and ethnic minorities including African American patients with AL amyloidosis given the higher pharmacokinetic (PK) exposure and hematologic toxicity rates (neutropenia, lymphopenia, thrombocytopenia and anemia). This study should characterize the exposure (including PK data), safety, and efficacy of daratumumab SC. |
| Janssen Biotech, Inc. | Tecvayli (teclistamab-cqyv) | PMR 4334-1: Conduct a randomized clinical trial in patients with relapsed or refractory multiple myeloma. The trial should enroll sufficient numbers of racial and ethnic minority patients and older patients (ages 65-74 and 75 and above) to enable an evaluation of teclistamab in a study population that better reflects the U.S. population of patients with multiple myeloma. Patients should be randomized to receive a teclistamab-based regimen compared to standard therapy for relapsed or refractory multiple myeloma. The primary endpoint should be progression-free survival and secondary endpoints should include overall survival, overall response rate, and duration of response. |
| Janssen Biotech, Inc. | Talvey (talquetamab-tgvs) | PMR 4473-1: Conduct a randomized clinical trial that evaluates the clinical efficacy and safety of talquetamab in patients with relapsed or refractory multiple myeloma. Patients should be randomized to receive a talquetamab-based regimen compared to standard therapy for relapsed or refractory multiple myeloma. The primary endpoint should be progression-free survival and secondary endpoints should include overall survival and overall response rate. The trial should enroll sufficient numbers of racial and ethnic minority patients and older patients (ages 65-74 years and 75 years and above) to enable an evaluation of talquetamab in a study population that reflects the U.S. population of patients with multiple myeloma. |
| Janssen Biotech, Inc. | Talvey (talquetamab-tgvs) | PMR 4473-2: Conduct a clinical trial to further characterize the serious risk of oral toxicity including severe stomatitis, mucositis, and weight loss, and potential mitigation measures, in patients receiving talquetamab 0.4 mg/kg weekly and talquetamab 0.8 mg/kg every two weeks. Evaluate incidence rates, time to onset, and outcomes including patient reported outcomes. Include investigation of associations and temporal relationships between incidence and severity of oral toxicity adverse events and other potential risk factors such as age, race and comorbidities, and impact of the mitigation measures evaluated. The study should enroll sufficient numbers of Black/African American patients to enable an evaluation of the serious risk of oral toxicity with talquetamab in this population. |
| KADMON PHARMACEUTICALS LLC | REZUROCK (Belumosudil) | PMC 4106-12: Conduct a clinical trial to assess the pharmacokinetics of belumosudil among U.S. racial and ethnic groups. This study should characterize the exposure (including PK data), efficacy, and safety of belumosudil. |
| KADMON PHARMACEUTICALS LLC | REZUROCK (Belumosudil) | PMR 4106-11: Conduct a clinical trial in a sufficient number of Black patients with chronic graft versus host disease to assess the risk of cardiac toxicities and further characterize Grade 3 toxicities including gastrointestinal and vascular disorders associated with the use of belumosudil. This study should characterize the exposure (including PK data), safety, and efficacy of belumosudil. |
| LOXO ONCOLOGY INC | Jaypirca (pirtobrutinib) | PMR 4389-1: Complete a randomized clinical trial to obtain data on the clinical efficacy and safety of pirtobrutinib in patients with mantle cell lymphoma. The trial should compare pirtobrutinib monotherapy to an investigator’s choice of approved BTK inhibitors in patients with mantle cell lymphoma. The primary endpoint should be progression-free survival as assessed by an independent review committee, with secondary endpoints that include overall survival and objective response rate. The trial should enroll a sufficiently representative study population to reflect the racial and ethnic diversity of the U.S. patient population with mantle cell lymphoma and allow for interpretation of the results in these patient populations. |
| Merck Sharp & Dohme LLC | Keytruda (pembrolizumab) | PMC 4531-2: Conduct an integrated analysis from ongoing, completed, or planned clinical trials and other potential data sources as appropriate enrolling a sufficient representation of older adults ages 75 years and older, and United States (U.S.) racial and ethnic minority patients that is reflective of the U.S. population of patients with NSCLC, to further characterize the efficacy and safety of pembrolizumab in combination with platinum-containing chemotherapy and pembrolizumab as a single agent in these patients. In the analysis, include a sufficient number of patients enrolled in the U.S., ages 75 years and older, and a sufficient number of racial and ethnic minorities reflective of the incidence of NSCLC in each subpopulation to allow for interpretation of the results. The analyses should support comparative efficacy and safety outcome analyses between the aforementioned populations and White, and younger patients. |
| MorphoSys US Inc. | Monjuvi (tafasitamab-cxix) | PMC 3904-2: Submit an integrated final report containing data from clinical trials (including data from the ongoing clinical development program), post-marketing reports, compassionate use/expanded access program, real-world data and other sources to further characterize the safety and efficacy of tafasitamab in combination with lenalidomide among U.S. racial and ethnic minority patients with DLBCL. |
| MorphoSys US Inc. | Monjuvi (tafasitamab-cxix) | PMR 3904-1: Submit the final report and datasets from a randomized, Phase 3 clinical trial to verify the clinical benefit of tafasitamab in patients with diffuse large B-cell lymphoma. The trial should include sufficient numbers of racial and ethnic minority patients to better reflect the U.S. patient population and allow for the interpretation of the results in these patient populations. Patients should be randomized to receive immunotherapy and/or chemotherapy with or without tafasitamab and lenalidomide. The primary endpoint should be progression-free survival, with secondary endpoints that include overall survival and objective response rate. |
| NOVARTIS PHARMACEUTICALS CORP | Vijoice (Alpelisib) | PMR 4260-1: Conduct a multiregional clinical trial to verify and describe the clinical benefit of alpelisib film-coated tablets, through more precise estimation of confirmed objective response rate and mature response duration per blinded independent review, in adult and pediatric patients 2 years of age and older with PIK3CA-Related Overgrowth Spectrum (PROS), including those with severe manifestations of PROS. Responding patients will be followed for at least 36 months from the onset of response, or until disease progression, whichever comes first. Evaluate a sufficient number of patients to characterize response rate and durability of response by PIK3CA mutation type (frequent hotspot mutations vs. other less frequent mutations), PROS subtype, and age (2 – 5 years, 6 – 11 years, 12 – 17 years, = 18 years). Include patient narratives and additional outcomes measures (such as clinical outcomes assessments) to support the assessment of clinical benefit in the study report. The distribution of race and ethnicity in the patient population studied should be sufficiently reflective of the U.S. patient population to support generalizability of results to U.S. patients with PROS. |
| ONYX PHARMACEUTICALS INC A WHOLLY OWNED SUB OF AMGEN INC | Kyprolis (Carfilzomib) | PMC 4183-1: Conduct an integrated study analysis containing data from clinical trials, post-marketing reports, compassionate use/expanded access programs, real-world evidence, and other sources to further characterize the safety and efficacy of daratumumab (SC) in combination with carfilzomib and dexamethasone among U.S. racial and ethnic minority patients with multiple myeloma. |
| ONYX PHARMACEUTICALS INC A WHOLLY OWNED SUB OF AMGEN INC | Kyprolis (Carfilzomib) | PMC 4279-2: Conduct an integrated analysis that contains data from clinical trials, postmarketing reports, compassionate use/expanded access programs, realworld evidence, and other sources to further characterize the safety and efficacy of carfilzomib in combination with isatuximab and dexamethasone (Isa-Kd) among U.S. racial and ethnic minority patients with multiple myeloma. |
| OTSUKA PHARMACEUTICAL CO LTD | JYNARQUE (tolvaptan) | PMR 3384-1: Conduct a prospective cohort study of patients enrolled in the Jynarque (tolvaptan) Risk Evaluation and Mitigation Strategies (REMS) registry, with the primary objective of determining the incidence rate of severe (fatal and potentially fatal) drug induced liver injury (DILI). The incidence rate should be compared to that observed in the development program (in TEMPO and REPRISE trials). Incidence rates should be stratified by important risk factors for DILI which at a minimum should include: age, gender, race, alcohol use, cumulative dose of tolvaptan and duration of tolvaptan use. |
| Pfizer Inc. | Elrexfio (elranatamab-bcmm) | PMC 4476-2: Conduct an integrated analysis of data from clinical trials to further characterize the efficacy, pharmacokinetics, pharmacodynamics, and safety of elranatamab among U.S. racial and ethnic minority patients with multiple myeloma. The population should be representative of the U.S. population of patients with multiple myeloma, including racial and ethnic diversity, and allow for interpretation of the results in these populations. |
| Regeneron Pharmaceuticals, Inc. | LIBTAYO (cemiplimab-rwlc) | PMC 4349-2: Conduct an integrated analysis containing data from ongoing or planned clinical trials and other data sources such as post-marketing reports, real-world evidence and other sources to further characterize the safety and efficacy of cemiplimab-rwlc in combination with platinum-doublet chemotherapy in older adults (e.g., ages 75 years and older), females, and racial and ethnic minority patients that is reflective of the U.S. population of patients with NSCLC. In the analysis, include a sufficient number of patients enrolled in the U.S., ages 75 years and older, females, and a sufficient number of racial and ethnic minorities reflective of the incidence of NSCLC in each subpopulation to allow for interpretation of the results. The analyses should support comparative safety and efficacy outcome analyses between the aforementioned populations and White, male, and younger patients. |
| sanofi-aventis U.S. LLC | Sarclisa (isatuximab-irfc) | PMC 4040-2: Submit a final report containing data from clinical trials, post-marketing reports, compassionate use/expanded access programs, real-world evidence, and other sources to further characterize the safety and efficacy of isatuximab in combination with carfilzomib and dexamethasone (Isa-Kd) among U.S. racial and ethnic minority patients with multiple myeloma. |
| SEAGEN INC | Tukysa (tucatinib) | PMR 4388-1: Conduct a randomized clinical trial to obtain data on the clinical efficacy of tucatinib for patients with RAS wild type, HER2-positive, unresectable or metastatic colorectal carcinoma. The trial should compare tucatinib in combination with trastuzumab with the standard of care in patients with RAS wild type, HER2-positive, unresectable or metastatic colorectal carcinoma. The primary endpoint should be progression-free survival (PFS) per blinded assessment or overall survival. The trial should enroll a sufficiently representative study population to reflect the racial and ethnic diversity of the U.S. patient population with RAS wild type, HER2-positive, unresectable or metastatic colorectal carcinoma and allow for interpretation of the results across this representative study population. |
| STEMLINE THERAPEUTICS INC | Orserdu (elacestrant) | PMC 4394-2: Conduct an integrated analysis containing data from clinical trials and other data sources such as post-marketing reports, real-world evidence and other sources to further characterize the safety and efficacy of elacestrant in patients from racial and ethnic minority groups. The analyses should support comparative safety and efficacy outcome analyses between the aforementioned populations and White patients. |
| TAIHO ONCOLOGY INC | Lytgobi (Futibatinib) | PMR 4345-1: Conduct a randomized clinical trial comparing dosages of futibatinib 16 mg and 20 mg once daily to verify and describe the clinical benefit of futibatinib in patients with advanced or metastatic cholangiocarcinoma harboring an FGFR2 gene fusion or other rearrangement. The overall response rate and duration of response should be assessed by a blinded independent review. The study should also evaluate other clinical outcomes that denote clinical benefit, such as patient reported outcomes. This study should enroll a minimum of 120 patients and all responders should have a minimum of 6 months from the date of initial response (or until disease progression, whichever comes first). Ensure that racial and ethnic minorities are adequately represented in the trial population, at a minimum, proportional to the prevalence of FGFR2 alterations in these subgroups in the US population. |
| TAKEDA PHARMACEUTICALS USA INC | EXKIVITY (mobocertinib) | PMC 4148-7: Conduct an analysis containing data from clinical trials enrolling a sufficient representation of U.S. racial and ethnic minorities, including Black or African American patients, that is reflective of the U.S. population of patients with EGFR exon 20 insertion-mutated NSCLC to further characterize the safety and efficacy of mobocertinib in Black or African American patients with EGFR exon 20 insertion-mutated NSCLC. |
| TAKEDA PHARMACEUTICALS USA INC | Fruzaqla (fruquintinib) | PMC 4544-1: Conduct a clinical study to further characterize the clinical effects of fruquintinib, including pharmacokinetics (PK), activity, blood pressure assessments, and safety events of palmar-plantar erythrodysesthesia in an underrepresented minority population. |